Содержание
Перечень нормативных документов для производственного контроля
Представленный ниже перечень нормативных документов, поможет вам решить вопросы, возникающие при организации и осуществлении производственного контроля за соблюдением санитарных правил.
| Федеральный закон от 30.03.1999 № 52-ФЗ с изменениями от 29 июля 2017 года | О санитарно-эпидемиологическом благополучии населения | Скачать |
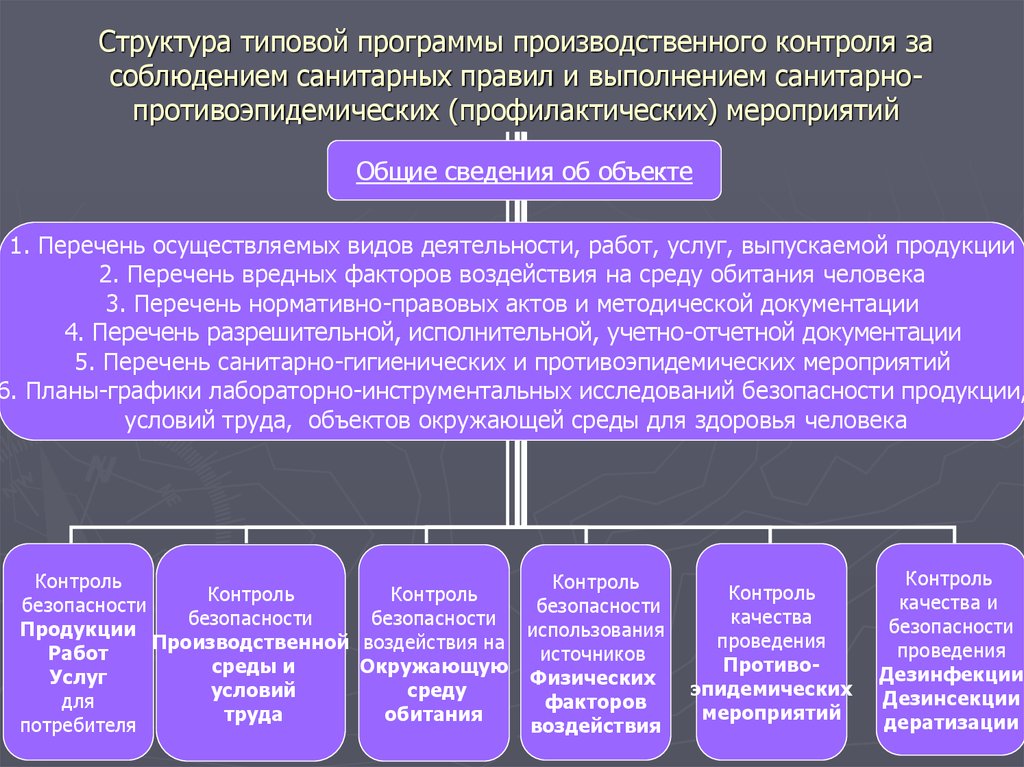
| СП 1.1.1058-01 с изменениями от 27 марта 2007 года | Организация и проведение производственного контроля за соблюдением санитарных правил и выполнением санитарно-противоэпидемических (профилактических) мероприятий | Скачать |
| Федеральный закон от 30.12.2001 № 195-ФЗ Кодекс РФ от 30.12.2001 № 195-ФЗ (статья 6.3) с изменениями от 29 июля 2017 года | Кодекс Российской Федерации об административных правонарушениях | Скачать |
| Письмо Роспотребнадзора от 15 февраля 2012 года № 01/1350-12-32 | О действии СП 1. 1.1058-01 1.1058-01 | Скачать |
| Письмо Роспотребнадзора от 13.04.2009 № 014801-9-32 | О типовых программах производственного контроля | Скачать |
| Приказ Минздравсоцразвития России от 12.04.2011 № 302н с изменениями от 5 декабря 2014 года | Об утверждении перечней вредных и (или) опасных производственных факторов иработ, при выполнении которых проводятся предварительные и периодические медицинские осмотры (обследования), и Порядка проведения предварительных и периодических медицинских осмотров (обследований) работников, занятых на тяжелых работах и на работах с вредными и (или) опасными условиями труда | Скачать |
| P 2.2.2006-05 | Руководство по гигиенической оценке факторов рабочей среды и трудового процесса. Критерии и классификация условий труда | Скачать |
| ГН 2.1.6.1338-03 с изменениями от 12 января 2015 года | Гигиенические нормативы. Предельно допустимые концентрации (ПДК) загрязняющих веществ в атмосферном воздухе населенных мест | Скачать |
ГН 2. 2.5.1313-03 с изменениями от 16 сентября 2013 года 2.5.1313-03 с изменениями от 16 сентября 2013 года | Гигиенические нормативы. Предельно допустимые концентрации (ПДК) вредных веществ в воздухе рабочей зоны | Скачать |
| СанПиН 2.1.4.1074-01 с изменениями от 28 июня 2010 года | Санитарно-эпидемиологические правила и нормативы. Питьевая вода. Гигиенические требования к качеству воды централизованных систем питьевого водоснабжения. Контроль качества. Гигиенические требования к обеспечению безопасности систем горячего водоснабжения | Скачать |
| СанПиН 2.2.0.555-96 | Санитарно-эпидемиологические правила и нормативы. Гигиенические требования к условиям труда женщин | Скачать |
| СанПиН 2.2.2.540-96 | Санитарно-эпидемиологические правила и нормативы. Гигиенические требования к ручным инструментам и организации работ | Скачать |
| СанПиН 2.2.4.548-96 | Санитарно-эпидемиологические правила и нормативы. Гигиенические требования к аэроионному составу воздуха производственных и общественных помещений | Скачать |
СанПиН 2. 2.4.3359-16 введены с 1 января 2017 года 2.4.3359-16 введены с 1 января 2017 года | Санитарно-эпидемиологические требования к физическим факторам на рабочих местах | Скачать |
| СанПиН 2.2.4.1191-03 с изменениями от 2 марта 2009 года | УТРАТИЛИ СИЛУ (опубликованы справочно) Электромагнитные поля в производственных условиях | Скачать |
| СанПиН 2.2.4.1294-03 | Гигиенические требования к аэроионному составу воздуха производственных и общественных помещений | Скачать |
| СанПиН 2.2.12.1.1.1278-03 с изменениями от 15 марта 2010 года | Санитарно-эпидемиологические правила и нормативы. Гигиенические требования к естественному, искусственному и совмещенному освещению жилых и общественных зданий | Скачать |
| СанПиН 2.3.2.1078-01 с изменениями от 6 июля 2011 года | Санитарно-эпидемиологические правила и нормативы. Гигиенические требования безопасности и пищевой ценности пищевых продуктов | Скачать |
СанПиН № 2. 2.22.4.1340-03 с изменениями от 21 июня 2016 года 2.22.4.1340-03 с изменениями от 21 июня 2016 года | Гигиенические требования к персональным электронно-вычислительным машинам и организации работы | Скачать |
| СН 2.2.42.1.8.562-96 | Санитарные нормы. Шум на рабочих местах, в помещениях жилых, общественных зданий и на территории жилой застройки | Скачать |
| СН 2.2.42.1.8.566-96 | Санитарные нормы. Производственная вибрация, вибрация в помещениях жилых и общественных зданий | Скачать |
| СП 2.2.2.1327-03 | Санитарно-эпидемиологические правила. Гигиенические требования к организации технологических процессов, производственному оборудованию и рабочему инструменту | Скачать |
| СП 44.13330.2011 | Свод правил. Административные и бытовые здания | Скачать |
| СП 52.13330.2011 | Свод правил. Естественное и искусственное освещение | Скачать |
Санитарно-промышленная лаборатория для целей производственного контроля.
Отчет о СОУТ
Карта спецоценки
Оценка факторов
Лист карты СОУТ
Подпись эксперта
Цена
© 2004-2022 Центр экспертизы условий труда – ЦЭУТ. Все права защищены.
Новости: Нормативные документы | АО НПО «Техкранэнерго» Нижегородский филиал
- Все
- Наши новости
- Ростехнадзор
- Нормативные документы
- Обучение/охрана труда
- СРО
- Страхование ОПО
- Лифты
- Выставки
- Прочее
« 1 2 3 4 5 … 38 »
23.09.2022 редакция от 27.09.2022
Новости портала regulation.gov.ru: Проекты НПА, разработанные Ростехнадзором в августе-сентябре 2022 года
О внесении изменений в технический регламент Таможенного союза «О безопасности оборудования, работающего под избыточным давлением» (ТР ТС 032/2013)… подробнее
02.09.2022 редакция от 01.09.2022
С 1 сентября 2022 года изменилось много нормативных документов
Приводим обзор изменений нормативки. .. подробнее
.. подробнее
01.09.2022 редакция от 31.08.2022
Перечень профессиональных стандартов, связанных с промышленной безопасностью, актуальных в 2022 году
Актуализировали Реестр профессиональных стандартов — 1489 шт. на 25.08.2022…… подробнее
29.07.2022 редакция от 31.08.2022
Отсрочена отмена актов Госгортехнадзора России по сварочному производству
перенесена с 01.09.2022 на 01.03.2023… подробнее
28.07.2022 редакция от 27.07.2022
Новости портала regulation.gov.ru: Проекты НПА, разработанные Ростехнадзором в июле 2022 года
Выйдут обновленные правила аттестации в области промышленной безопасности, по вопросам безопасности гидротехнических сооружений, безопасности в сфере электроэнергетики… подробнее
22.07.2022
Регуляторная «гильотина разъяснений» затронет руководства по безопасности, методические рекомендации
до конца года по 45 ведомствам подобные документы планируется сократить и систематизировать. .. подробнее
.. подробнее
19.07.2022 редакция от 21.07.2022
Новые ГОСТы в промышленной безопасности 2022 года
Мы следим за новыми Гостами, связанными с нашими видами работ и вообще с промышленной безопасностью, и обновляем на этой странице …… подробнее
19.07.2022 редакция от 19.09.2022
Ростехнадзор разъяснил статус своих писем с ответами на вопросы
территориальные органы Ростехнадзора, обязаны руководствоваться такими разъяснениями в соответствующей правоприменительной деятельности… подробнее
08.07.2022 редакция от 06.07.2022
Ростехнадзор разъясняет: Пуск газа или ввод в эксплуатацию газовых объектов
По какой форме составлять Акта приемки законченного строительства объекта сети газораспределения (газопотребления)?… подробнее
06.07.2022 редакция от 29.06.2022
Ростехнадзор разъясняет: Вопросы эксплуатации складов нефти и нефтепродуктов (нефтебаз)
Возможен ли замер уровня нефтепродуктов в резервуарах через люк на крыше резервуара замерной лентой или рейкой?
Каким документом регламентируется периодичность зачистки мазутных баков?
Письмо Ростехнадзора от 16. 06.2021 N 14-00-09/1535 «О результатах рассмотрения обращения»
06.2021 N 14-00-09/1535 «О результатах рассмотрения обращения»
Прошу сообщить нормативные акты, предписывающие гидравлический и механический расчет технологических трубопроводов, подбор насосных агрегатов исходя из расчетных данных для складов нефти и нефтепродуктов…. подробнее
05.07.2022 редакция от 15.07.2022
Новые ГОСТы в промышленной безопасности, которые вводятся с 2022 года
Мы следим за новыми Гостами, связанными с нашими видами работ и вообще с промышленной безопасностью, и обновляем на этой странице …… подробнее
05.07.2022 редакция от 29.06.2022
Ростехнадзор разъясняет: Сведения, характеризующие ОПО
Необходима ли перерегистрация ОПО при изменении названия эксплуатирующей организации?
Что может относиться к соответствующим документам, подтверждающим наличие оснований для внесения изменений в сведения о характеристиках ОПО?
Допускается ли представление документов, подтверждающих наличие оснований для внесения изменений, только в виде электронных копий документов на съемном электронном носителе?. .. подробнее
.. подробнее
04.07.2022 редакция от 30.06.2022
Ростехнадзор разъясняет: Дымовые трубы
Обязательно ли составление паспорта на промышленную металлическую дымовую трубу?… подробнее
30.06.2022 редакция от 29.06.2022
Ростехнадзор разъясняет: Консервация ОПО
Если опасный производственный объект находится на консервации, нужна экспертиза промышленной безопасности?… подробнее
29.06.2022
Ростехнадзор разъясняет: Исключение ОПО из реестра
Как исключить объект из реестра опасных производственных объектов в связи с расторжением договора на эксплуатацию?… подробнее
27.06.2022 редакция от 26.06.2022
Ростехнадзор разъясняет: О добровольной сертификации сварочного производства
Проведение аттестации сварочного производства органами систем добровольной сертификации законодательством не предусмотрено…. подробнее
14.06.2022 редакция от 10.06.2022
В 2023 году заключения ЭПБ будут содержать однозначный вывод
исключается вывод — «не в полной мере соответствует требованиям промышленной безопасности». .. подробнее
.. подробнее
07.06.2022
Опубликованы Правила по эксплуатации технологических трубопроводов на ОПО
Вступают в силу с сентября этого года…. подробнее
01.06.2022
Ростехнадзор разработал новые руководства по безопасности
Анализ опасностей и оценки риска аварий на опасных производственных объектах и методика моделирования распространения аварийных выбросов опасных веществ… подробнее
01.06.2022
МЧС России наращивает эффективность новой системы надзора
Контрольная деятельность строится на принципах риск-ориентированного подхода. Каждому зданию присваивается индивидуальная категория риска с учетом его пожарно-технических характеристик и добросовестности собственников…. подробнее
« 1 2 3 4 5 … 38 »
Нормативно-правовые распорядительные акты
Нормативно-правовые распорядительные акты
Федеральные, региональные муниципальные акты
Федеральный закон от 30. 03.1999 N 52-ФЗ (ред. от 13.07.2020) «О санитарно-эпидемиологическом благополучии населения»
03.1999 N 52-ФЗ (ред. от 13.07.2020) «О санитарно-эпидемиологическом благополучии населения»
Федеральный закон от 02.01.2000 N 29-ФЗ (ред. от 13.07.2020) «О качестве и безопасности пищевых продуктов»
Технический регламент таможенного союза ТР ТС 021/2011 «О безопасности пищевой продукции»
Технический регламент таможенного союза ТР ТС 022/2011 «Пищевая продукция в части ее маркировки»
СанПиН 2.3.2.1078-01. «Гигиенические требования безопасности и пищевой ценности пищевых продуктов»
СанПиН 2.3.2.1324-03. «Гигиенические требования к срокам годности и условиям хранения пищевых продуктов»
СП 1.1.1058-01. Организация проведения производственного контроля за соблюдением санитарных правил и выполнением санитарно- противоэпидемических (профилактических) мероприятий.
СанПиН 2.3.2.4.3590-20 «Санитарно-эпидемиологические требования к организации питания обучающихся в общеобразовательных учреждениях, учреждениях начального и среднего профессионального образования» (далее — СанПиН 2. 4.5.2409-08).
4.5.2409-08).
СанПиН 2.3/2.4.3590-20 «Санитарно-эпидемиологические требования к организации общественного питания населения». Регистрационный № 60833 от 11 ноября 2020 г.
Методические рекомендации МР 2.4.0162-19 «Особенности организации питания детей, страдающих сахарным диабетом и иными заболеваниями, сопровождающимися ограничениями в питании (в образовательных и оздоровительных организациях)» (далее — МР 2.4.0162-19)
Методические рекомендации МР 2.4.0172-20 «Рекомендации по организации питания обучающихся общеобразовательных организаций»
Методические рекомендации МР 2.4.0181-20 «Родительский контроль за организацией горячего питания детей в общеобразовательных организациях»
Приказ Минздравсоцразвития России N 213н, Минобрнауки России N 178 от 11.03.2012 «Об утверждении методических рекомендаций по организации питания обучающихся и воспитанников образовательных учреждений»
Приказ Министерства образования и науки Пермского края от 14.08.2020 № 26-01-05-238 «О повышении эффективности системы контроля качества предоставления питания обучающимся муниципальных общеобразовательных организаций, расположенных на территории Пермского края, государственных образовательных организаций, находящихся в ведении Министерства образования и науки Пермского края»
Закон Пермской области от 09 сентября 1996 г. № 533-83 «О социальных гарантиях и мерах социальной поддержки семьи, материнства, отцовства и детства»
№ 533-83 «О социальных гарантиях и мерах социальной поддержки семьи, материнства, отцовства и детства»
Постановление Правительства Пермского края от 06 июля 2007 г. № 130-п «О предоставлении мер социальной поддержки малоимущим семьям, имеющим детей, и беременным женщинам»
Решение Пермской городской Думы от 27 ноября 2007 г. № 280 «О предоставлении бесплатного питания отдельным категориям обучающихся в муниципальных общеобразовательных учреждениях города Перми»
Решение Пермской городской Думы от 21 ноября 2017 г. № 228 «О предоставлении бесплатного двухразового питания учащимся с ограниченными возможностями здоровья, обучающимся в муниципальных общеобразовательных учреждениях города Перми, частных общеобразовательных организациях»
Постановление администрации г. Перми от 14.06.2018 N 391 (ред. от 01.02.2021) «Об утверждении Порядка предоставления бесплатного питания учащимся в общеобразовательных организациях города Перми»
Постановление администрации г. Перми от 14. 06.2018 N 391 «Об утверждении Порядка предоставления бесплатного питания учащимся в общеобразовательных организациях города Перми»
06.2018 N 391 «Об утверждении Порядка предоставления бесплатного питания учащимся в общеобразовательных организациях города Перми»
Приказ начальника департамента образования администрации города Перми от 24.09.2019 № СЭД-059-08-01-09-918 «Об утверждении положения по организации питания в муниципальных общеобразовательных учреждениях города Перми»
Приказ начальника департамента образования от 27.08.2021 № 059-08-01-09-925 «Об организации питания в подведомственных муниципальных общеобразовательных учреждениях в 2021-2022 учебном году»
Приказ начальника департамента образования администрации города Перми 27.08.2021 №059-08-01-09-922 «О внесении изменений в Положение об организации питания в муниципальных учреждениях города Перми, утвержденное приказом начальника департамента образования администрации города Перми от 24.09.2019 №059-08-01-09-918»
Письмо Министерства образования и науки Пермского края о размере денежных норм на питание с 1 января 2022 года
Локальные акты:
Положение об общественной комиссии
Положение о бракеражной комиссии
Положение об организации питания учащихся школы
Положение о школьной карте
Приказ об организации питания учащихся школы
Программа производственного контроля организации питания
Программа административного контроля организации питания
Приказ об организации деятельности по формированию навыков и культуры здорового питания обучающихся
Приказ о создании общественной комиссии по осуществлению контроля за питанием обучающихся
План деятельности общественной комиссии за питанием МАОУ «СОШ №42» г. Перми на 2022-2023 учебный год
Перми на 2022-2023 учебный год
План проведения проверок общественной комиссией за питанием МАОУ «СОШ №42» г.Перми на 2022-2023 учебный год
Состав бракеражной комиссии
Состав комиссии по общественному контролю
Приказ об утверждении новых редакций документов по организации питания обучающихся
График предоставления питания
Режим работы столовой
Бланки заявлений на предоставление бесплатного питания
Производственный контроль | ЮгТрансТест
Южно – региональный центр «ЮгТрансТест» с 2003 года оказывает услуги по проведению производственного контроля и разработке программы производственного контроля в соответствии с действующим законодательством.
Уточнить стоимость производственного контроля вы можете по телефону: 8-800-200-61-77
Проведение производственного контроля осуществляется в соответствии с требованиями Федерального закона от 30 марта 1999 года № 52-Ф3 “О санитарно-эпидемиологическом благополучии населения” (ред. от 23.06.2014) и СП 2.2.3670-20 “Санитарно-эпидемиологические требования к условиям труда” (далее – СП 2.2.3670-20).
от 23.06.2014) и СП 2.2.3670-20 “Санитарно-эпидемиологические требования к условиям труда” (далее – СП 2.2.3670-20).
Цель производственного контроля: обеспечение безопасности и (или) безвредности для человека и среды обитания вредного влияния объектов производственного контроля путем должного выполнения санитарных правил, санитарно-противоэпидемических (профилактических) мероприятий, организации и осуществления контроля за их соблюдением. На каждом предприятии должны быть обеспечены безопасные для человека условий труда. Стоимость производственного контроля зависит от масштаба организации (численность сотрудников, количество основных фондов, экстерриториальные особенности и т.п.).
Согласно Федеральному закону №52-ФЗ юридические лица, осуществляющие предпринимательскую деятельность, и индивидуальные предприниматели обязаны следить за соблюдением санитарно-эпидемиологических норм на рабочих местах сотрудников, а также выполнять предписания контролирующих органов, выполняющих свои функции в данной сфере. На каждом предприятии должен быть документ, регламентирующий контроль санитарно-гигиенических норм этого предприятия – программа производственного контроля.
На каждом предприятии должен быть документ, регламентирующий контроль санитарно-гигиенических норм этого предприятия – программа производственного контроля.
Порядок разработки и основные разделы программы производственного контроля определены в Методических рекомендациях МР 2.2.0244-21 “Методические рекомендации по обеспечению санитарно-эпидемиологических требований к условиям труда” (утв. Федеральной службой по надзору в сфере защиты прав потребителей и благополучия человека 17 мая 2021 г.) и Санитарных правилах СП 1.1.1058–01 «Организация и проведение производственного контроля за соблюдением санитарных правил и выполнением санитарно-противоэпидемических (профилактических) мероприятий. Санитарные правила» (в ред. Изменений и дополнений № 1 (СП 1.1.2193–07), утв. Постановлением Главного государственного санитарного врача РФ от 27.03.2007 № 13). Проведение производственного контроля обязательно для всех предприятий РФ.i
Производственный контроль за условиями труда обязаны проводить все юридические лица и индивидуальные предприниматели в соответствии с осуществляемой ими деятельностью, а так же обеспечивать контроль за соблюдением санитарных правил, гигиенических нормативов и выполнением санитарно-профилактических мероприятий, направленных на снижение рисков для здоровья человека.
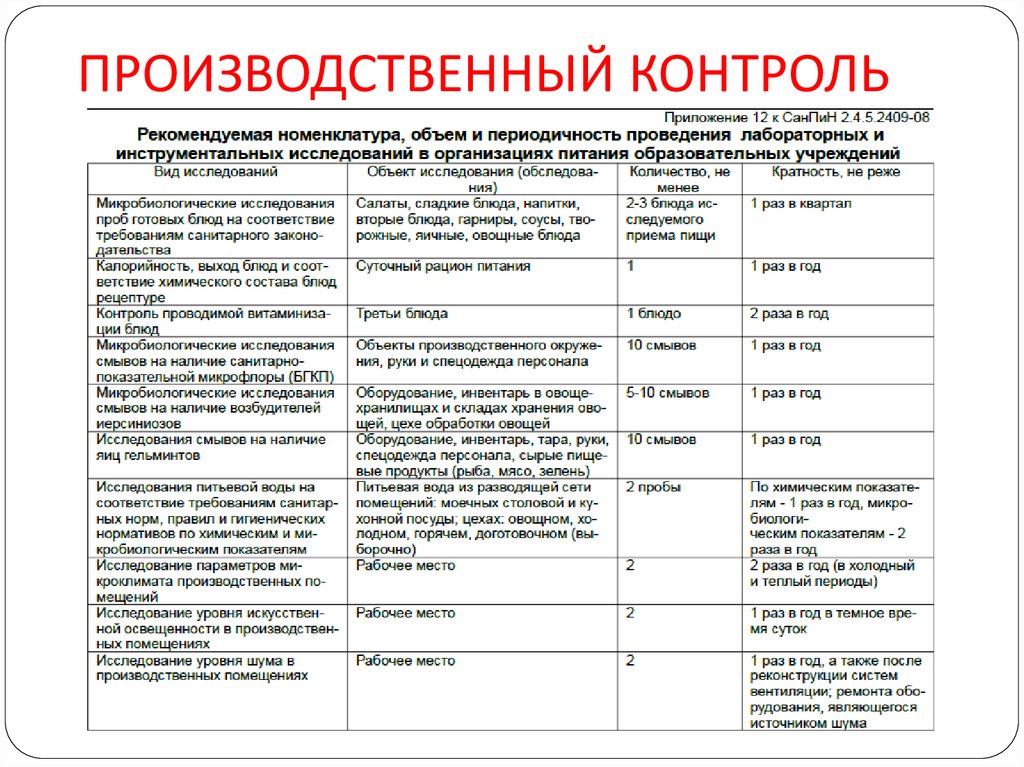
Объем и периодичность исследований определяются действующими нормативными документами РФ для исследуемого параметра, а количество контролируемых параметров на каждом рабочем месте определяется в соответствии с производственным процессом.
Наша аккредитованная лаборатория оказывает услуги по проведению производственного контроля и поможет в разработке программы производственного контроля, учитывая специфику и сферу деятельности Вашей компании, а также в дальнейшем проведении измерений по необходимым факторам производственного процесса согласно области аккредитации. Сотрудники нашей лаборатории организуют и проведут мероприятия, которые являются необходимыми для исполнения программы производственного контроля. Стоимость осуществления измерений зависит от факторов рабочей среды.
Согласно ст. 12 Федерального закона № 426-ФЗ результаты исследований и измерений вредных и (или) опасных производственных факторов, которые были проведены в рамках производственного контроля за условиями труда, могут использоваться в качестве результатов исследований при проведении специальной оценки условий труда, если исследования проводились лабораторией, аккредитованной в Федеральном органе по аккредитации.
В соответствии с требованиями СП 2.2.3670-20 (пп.2.4) лабораторные исследования и испытания организуются хозяйствующим субъектом и проводятся испытательной лабораторией (центром), принадлежащей хозяйствующему субъекту, или иной лабораторией (центром), аккредитованной в соответствии с законодательством Российской Федерации об аккредитации в национальной системе аккредитации.
Испытательная лаборатория ООО “ЮРЦ “ЮгТрансТест” одна из немногих, аккредитованных в Федеральном органе по аккредитации.
Заключив договор на проведение производственного контроля, Вы экономите на услугах по проведению специальной оценки условий труда.
Внимание!!!
Если производственный контроль не проводился, то это является нарушением пп. 1.4 СП 2.2.3670-20 “Санитарно-эпидемиологические требования к условиям труда” и пп. 2.3, 2.4 (а), 2.5, 2.7 СП 1.1.1058-01 “Организация и проведение производственного контроля за соблюдением Санитарных правил и выполнение санитарно-противоэпидемиологических (профилактических) мероприятий”.
Нормативные документы | CCTS
Основные документы обычно называют нормативными документами. Руководство ICH GCP определяет основные документы как «те документы, которые по отдельности и в совокупности позволяют оценить проведение клинического исследования и качество полученных данных. Эти документы служат для демонстрации соответствия исследователя, спонсора и монитора стандартам надлежащей клинической практики и всем применимым нормативным требованиям».
В начале любого исследования должен быть создан основной файл исследования, который должен поддерживаться на протяжении всего исследования. Этот файл помогает в управлении исследованием и часто является элементом, который проверяется наблюдателями за исследованием и должностными лицами FDA. Если не указано иное, копии всех документов должны быть помещены в мастер-файл на сайте и у спонсора исследования или контрактной исследовательской организации (CRO).
- ICH E6 Надлежащая клиническая практика (GCP)
В следующих разделах описаны необходимые документы в соответствии с Руководством ICH E6 для промышленности: Надлежащая клиническая практика (GCP). Обратите внимание, что все исследования на людях должны соответствовать GCP ICH независимо от типа клинического исследования.
Обратите внимание, что все исследования на людях должны соответствовать GCP ICH независимо от типа клинического исследования.
- Брошюра исследователя (IB)
- Для документального подтверждения того, что релевантная и актуальная научная информация об исследуемом продукте была предоставлена исследователю
- Обычно в ИБ имеется страница для подписи, которую должен подписать главный исследователь (PI) и вернуть спонсору исследования.
- Копию страницы подписи IB следует сохранить
- Когда выпускаются обновленные IB, они должны быть сохранены соответствующим образом. Устаревшие IB не следует удалять из файла.
- Форма FDA 1572
- Для документального подтверждения согласия исследователя предоставить определенную информацию спонсору и заверить, что он/она будет соблюдать правила FDA, касающиеся проведения клинического исследования исследуемого лекарственного средства или биологического препарата.

- Скачать последнюю версию формы 1572
- Для документального подтверждения согласия исследователя предоставить определенную информацию спонсору и заверить, что он/она будет соблюдать правила FDA, касающиеся проведения клинического исследования исследуемого лекарственного средства или биологического препарата.
- Журнал делегирования обязанностей
- Задокументировать делегирование исследователем определенных обязанностей исследовательскому персоналу для проведения клинического испытания.
- и поправки
- Обычно в протоколе/поправке есть страница для подписи, которую должен подписать главный исследователь (PI) и вернуть спонсору исследования
- Копию страницы подписи протокола/поправки следует сохранить
- Когда выпускаются поправки, они должны быть соответственно подшиты. Устаревшие протоколы не следует удалять из файла
- Информация, предоставленная участнику исследования
- Информированное согласие
- Для документирования процесса информированного согласия
- Этот документ должен быть одобрен IRB, прежде чем его можно будет использовать
- Получить дополнительную информацию о процессе получения информированного согласия
- Другая письменная информация
- Документально подтвердить, что участники исследования дали свое полностью информированное согласие
- Объявление о наборе персонала
- Документально подтвердить, что меры по найму являются надлежащими, а не принудительными
- Этот документ должен быть одобрен IRB, прежде чем его можно будет использовать
- Получить больше информации о найме
- Информированное согласие
- (FDF)
- Для документирования финансового соглашения между исследователем/организацией и спонсором исследования
- Форма FDF должна быть заполнена и подписана каждым сыщиком. Оригинал должен быть возвращен спонсору исследования, а копия сохранена
- Новый FDF должен быть заполнен каждый раз, когда происходит изменение финансового положения исследователя (например, покупка акций и т. д.)
- Генеральное соглашение о клинических испытаниях (MCTA)
- Для документирования соглашений между PI/учреждением и спонсором исследования и/или CRO
- Одобрение IRB
- Для документального подтверждения того, что исследование было одобрено IRB.
- Утверждение будет предоставлено для нескольких элементов, включая, помимо прочего:
- Протокол
- Документ информированного согласия
- Объявления о наборе персонала и другая письменная информация, которая должна быть предоставлена участнику исследования
- Онлайн-портал для поведенческих и социальных наук, биомедицинских наук и онкологических IRB
- Онлайн-портал для первоначального обзора исследований, спонсируемых и инициируемых отраслью
- Реестр IRB
- Документально подтвердить, что IRB создан в соответствии с Надлежащей клинической практикой
- ORRP публикует свои списки IRB, однако номер Федеральной страховой компании (FWA) может быть предоставлен вместо списка IRB.
- ФБД №: 00006378
- Переписка IRB
- Документировать всю переписку между исследовательским центром и IRB.
- Сюда следует включить всю переписку по электронной почте.
- Биографические справки (CV)
- Для документирования квалификации и права на проведение исследования и/или обеспечение медицинского наблюдения за участниками исследования
- Требуется для каждого исследователя, участвующего в исследовании
- CV должны быть актуальными и обновляться каждые два года, указывать связь исследователя с учреждением, в котором проводится исследование, и отображать адрес, указанный в 1572 (если применимо)
- Устаревшие резюме нельзя удалять из файла
- ГОРЯЧИЙ СОВЕТ: Хорошо бы хранить мастер-файл с резюме следователя, чтобы они были доступны для всех исследований. Это хорошо работает для медицинских лицензий, а также лабораторных сертификатов.
- Медицинская лицензия
- Для документирования квалификации и права на проведение исследования и/или обеспечение медицинского наблюдения за участниками исследования
- Требуется для каждого врача-исследователя, участвующего в исследовании
- Все лицензии должны быть действующими
- Не следует удалять лицензии с истекшим сроком действия из файла
- Записи о тренировках
- Документально подтвердить, что исследовательский персонал прошел соответствующую подготовку для выполнения протокола.
- Обучение может включать ознакомительный визит на объект или участие в совещании следователя.
- Запись об обучении должна отражать соответствующую подготовку всего исследовательского персонала.
- Это может включать обучение сбору образцов, обращению с ними и их хранению.
- Документально подтвердить, что исследовательский персонал прошел соответствующую подготовку для выполнения протокола.
- Лабораторная сертификация или аккредитация
- Документально подтвердить компетентность учреждения проводить требуемые испытания и поддерживать надежность результатов
- Многие спонсоры требуют сертификации как CAP, так и CLIA; однако это очень зависит от спонсора и протокола
- Сертификаты должны быть действующими
- Не следует удалять сертификаты с истекшим сроком действия из файла
- Лабораторные нормальные значения
- Документировать нормальные значения и/или диапазоны тестов
- Отслеживание отчетов о посещениях
- Для документирования завершенных мероприятий и решения любых проблем, отмеченных во время визита наблюдателя.
- Отчет о посещении объекта (досудебный мониторинг)
- Отчет об открытии Зоны или отчет о посещаемости собрания исследователей
- Отчеты о контрольных посещениях
- Отчет о заключительном посещении
- Для документирования завершенных мероприятий и решения любых проблем, отмеченных во время визита наблюдателя.
- Спонсорская переписка
- Документировать всю переписку между исследовательским центром и спонсором/CRO.
- Сюда следует включить всю переписку по электронной почте.
- Разная документация
- Образец этикетки, прикрепленной к контейнерам с исследуемым продуктом
- Инструкции по обращению с исследуемым продуктом и материалами, относящимися к испытаниям
- Записи об отгрузке исследуемых продуктов и материалов, связанных с испытаниями
- Сертификаты анализа отгруженной исследовательской продукции
- Процедуры декодирования слепых испытаний
- Исходные документы
- Для документирования существования субъекта и подтверждения достоверности собранных данных исследования.
- См. Ознакомительные поездки и управление документами
- Для документирования существования субъекта и подтверждения достоверности собранных данных исследования.
- Уведомление исследователя о серьезных нежелательных явлениях спонсору
- Уведомление инициатора исследования спонсору о серьезных нежелательных явлениях и соответствующие отчеты
- См. раздел о SAE
- Уведомление регулирующих органов о серьезных нежелательных явлениях спонсором
- Уведомление регулирующих органов и ЭСО спонсором и/или исследователем о неожиданных серьезных нежелательных явлениях
- См. раздел о SAE
- Журнал проверки субъекта
- Для документирования идентификации субъектов, прошедших досудебный скрининг
- Журнал идентификации субъекта
- Документально подтвердить, что исследователь/учреждение хранит конфиденциальный список имен всех испытуемых
- Позволяет следователю/учреждению раскрыть личность любого субъекта
- Журнал регистрации субъектов
- Для документирования хронологического набора субъектов по испытательному номеру
- Подписной лист
- Документировать подписи и инициалы всех сотрудников, участвующих в исследовании
- Журнал учета исследовательской продукции
- Для документального подтверждения того, что исследуемые продукты использовались в соответствии с протоколом
- См. раздел о SAE
- Документация об уничтожении исследуемого препарата
- Документировать уничтожение неиспользованных исследовательских продуктов спонсором или на месте
- См. раздел о SAE
Протокол
Форма раскрытия финансовой информации
 Оригинал должен быть возвращен спонсору исследования, а копия сохранена
Оригинал должен быть возвращен спонсору исследования, а копия сохранена

 раздел о SAE
раздел о SAEСтандарты контроля документов | Greenlight Guru
Компании, производящие медицинское оборудование, которые хотят продавать свою продукцию по всему миру, должны соблюдать стандарты контроля документации, установленные регулирующими органами, которые устанавливают требования к системе качества для каждого рынка.
Стандарты управления документами исторически в течение некоторого времени включались в нормативные документы по системам качества в различных отраслях промышленности. Исторически сложилось так, что организации, которые управляли большим объемом документов, часто нанимали менеджера по работе с документами, роль которого заключалась в управлении документами в соответствии с применимыми стандартами управления документами. Сегодня инновационные программные решения eQMS могут помочь компаниям автоматизировать или оптимизировать многие аспекты процесса контроля документов.
Сегодня инновационные программные решения eQMS могут помочь компаниям автоматизировать или оптимизировать многие аспекты процесса контроля документов.
Целью управления документацией является управление информацией, которая передается между соответствующими сторонами внутри и за пределами организации. Если оператору необходимо просмотреть СОП для определенного этапа производства, он должен иметь возможность своевременного доступа к правильной информации. Если информация должна быть изменена, должен быть предусмотрен процесс отслеживания и утверждения этих изменений. Как только происходит изменение, операторам необходим доступ к новому документу, а устаревшая версия должна быть удалена. Все эти процессы и функции облегчаются стандартами документооборота.
Правила контроля документов в QSR FDA
Хотя QSR FDA устанавливает требования к контролю документов, было бы неправильно называть эти требования «стандартом». Между стандартами и правилами существует важное различие: если вы не соответствуете стандарту, вы можете просто не иметь права продавать на этом рынке, в то время как несоблюдение правил может повлечь за собой финансовые или юридические санкции и , запрет на продажу товар.
Положения о контроле документации в FDA QSR требуют, чтобы компании, производящие медицинское оборудование, назначали ответственное лицо для проверки и утверждения документов, обеспечения доступности текущих документов, предотвращения непреднамеренного использования устаревших документов, ведения учета изменений документов и проведения повторных утверждений всякий раз, когда документ изменяется.
Стандарты управления документами: ISO 13485:2016
Наиболее актуальные стандарты документов на сегодняшний день основаны на ISO 13485:2016, ведущем международном стандарте систем качества медицинского оборудования. Компании, производящие медицинские устройства, которые хотят продавать продукцию на всей территории Европейской экономической зоны, должны соблюдать стандарты контроля документации ISO 13485: 2016, которые включают положения для:
- Проверка и утверждение документов
- Проверка, обновление и повторное утверждение документов
- Запись текущего статуса редакции и изменений в документах
- Обеспечение доступности соответствующих версий применимых документов в точках использования
- Обеспечение разборчивости и идентификации документов
- Контроль распространения и идентификации необходимых документов внешнего происхождения
- Предотвращение порчи или потери документов
- Предотвращение непреднамеренного использования устаревших документов
Взгляд в будущее: стандарты контроля документов в MDSAP
Мир регулирования медицинских устройств движется к новой стратегии регулирования, известной как Единая программа аудита медицинских устройств (MDSAP). Сотрудничающие страны в MDSAP включают Австралию, Бразилию, Канаду, Японию и Соединенные Штаты. Цель MDSAP — позволить компаниям, производящим медицинское оборудование, продавать продукцию на нескольких международных рынках на основе результатов одного аудита. В действующих стандартах документооборота MDSAP перечислены следующие требования к компаниям, производящим медицинское оборудование:
Сотрудничающие страны в MDSAP включают Австралию, Бразилию, Канаду, Японию и Соединенные Штаты. Цель MDSAP — позволить компаниям, производящим медицинское оборудование, продавать продукцию на нескольких международных рынках на основе результатов одного аудита. В действующих стандартах документооборота MDSAP перечислены следующие требования к компаниям, производящим медицинское оборудование:
- Утвердить документы на адекватность перед использованием
- Проверка и обновление по мере необходимости и повторное утверждение документов
- Убедитесь, что изменения и текущий статус редакции документов идентифицированы
- Убедитесь, что соответствующие версии применимых документов доступны в точках использования
- Убедитесь, что документы остаются разборчивыми и легко идентифицируемыми
- Убедиться, что документы внешнего происхождения, необходимые для планирования и функционирования СМК, идентифицированы, а их распространение контролируется
- Предотвращение непреднамеренного использования устаревших документов и применение к ним надлежащей идентификации, если они сохраняются для каких-либо целей
Компании, производящие медицинское оборудование, должны принять к сведению, что требования MDSAP в значительной степени основаны на требованиях ISO 13485:2016 и взяты из них. По мере того, как мы продвигаемся к принятию единой программы аудита для компаний, производящих медицинское оборудование, производители должны подготовиться к соблюдению стандартов контроля документации, изложенных в ISO 13485 и раннем руководстве MDSAP.
По мере того, как мы продвигаемся к принятию единой программы аудита для компаний, производящих медицинское оборудование, производители должны подготовиться к соблюдению стандартов контроля документации, изложенных в ISO 13485 и раннем руководстве MDSAP.
Greenlight Guru помогает производителям медицинского оборудования соответствовать стандартам контроля документации
Платформа программного обеспечения управления качеством Greenlight Guru гарантирует, что производители медицинского оборудования всегда могут получить доступ к своим самым последним СОП и документации по качеству в соответствующей точке использования, при этом автоматизируя записи об изменениях и предотвращение потери документов. Greenlight Guru позволяет менеджерам по качеству легко утверждать документы, повторно утверждать измененные документы и удалять устаревшие документы из обращения одним нажатием кнопки.
Greenlight Guru — это единственная система eQMS, разработанная для компаний, производящих медицинское оборудование, в виде готового решения. Платформа полностью совместима с самыми последними встроенными стандартами управления документами, что означает меньшее количество настроек и сокращение времени для производителей устройств, чтобы вывести свой продукт на рынок.
Платформа полностью совместима с самыми последними встроенными стандартами управления документами, что означает меньшее количество настроек и сокращение времени для производителей устройств, чтобы вывести свой продукт на рынок.
Ссылки по теме
- Корректирующие и предупреждающие действия
- FDA QSR
- Основная запись устройства
- 510k Представление
- 21 CFR часть 11
- ИСО 13485
Ищете комплексное решение QMS для продвижения успеха ваших устройств на рынке и интеграции ваших процессов обеспечения качества с усилиями по разработке продуктов? Нажмите здесь, чтобы получить краткий обзор программного обеспечения Greenlight Guru Medical Device QMS →
Контроль производственной документации — ресурсы продукта
Но не менее важным, чем сама документация, является то, как вы организуете и обновляете свои документы, чтобы вы всегда знали, какой документ является последним, а какие документы пересматриваются.
Одной из важных особенностей производственной системы является контроль документации, которая используется для производства продукта. Эта документация включает в себя все документы, используемые в производстве:
- Документы со спецификациями компонентов
- Чертежи нестандартных компонентов (деталь из листового металла, деталь из обработанной детали, деталь отлитой детали и т. д.)
- Документация по печатной плате
- И т. д.
- Сборочные чертежи и процедуры
- Процедуры испытаний
- Формы контрольного списка
- Формы FAT (заводские приемочные испытания)
- Документы контроля производственного процесса
- СОП — стандартные операционные процедуры компании (например, план контроля электростатического разряда, процедура комплектования)
- Документы СМК – система менеджмента качества
Эти документы имеют несколько важных особенностей помимо информации, которую они передают:
- Заголовок – для помощи в поиске документа
- Номер документа — уникальный код, который идентифицирует документ и отслеживает его через системы ERP и QMS
- Редакция документа — числовое или буквенное обозначение, идентифицирующее редакцию документа.
- Уполномочивающая подпись или другое указание на то, что документ одобрен для изготовления.

Информация в документе является целью документа, но метаданные документа, номер документа и редакция показывают, что информация, которая будет использоваться, является точно правильной версией информации. Если существует несколько ревизий процедуры сборки, скажем, ревизий A, B и C, для новых производственных сборок всегда используется самая последняя (C). Может быть только одна версия документа Rev C. Если вы должны спросить, есть ли у вас последняя версия документа Rev C, вы делаете это неправильно; более того, если назвать что-то «последней версией», это может вызвать проблемы в продакшене. Разработка может ссылаться на документ, который является «последней версией», в то время как у производства есть совершенно другая «последняя версия», которая содержит другую информацию, которая может повлиять на продукт. Контроль версий объединяет всех заинтересованных сторон, связанных с документом.
Контроль версий объединяет всех заинтересованных сторон, связанных с документом.
Инженерные модели
Почти для всех документов за документом стоит файл или база данных. Сборочный чертеж можно опубликовать в формате PDF (переносимый формат документа), но за этим PDF-файлом находится чертеж, связанный с твердотельной моделью продукта. Твердотельная модель (для механических параметрических программ САПР) представляет собой базу данных, содержащую определение сборки. Он не используется непосредственно в производстве, но также должен иметь контроль версий. Для электрических сборок, таких как печатная плата, существует графическое изображение (например, фотосхемы Gerber или программный файл, такой как ODB++), который является результатом, отправляемым поставщику для изготовления печатной платы и заполнения компонентов. За этим стоит база данных печатных плат, будь то в PADS, Eagle, OrCad, Altium или какой-либо другой программе компоновки печатных плат. Процедура сборки или тестирования может быть разработана в Microsoft Word; документ Word — это мастер-файл, а PDF — это выходной файл для производства.
Еще одной тонкостью управления инженерными моделями являются полномочия и ответственность за поддержание модели. По мере реализации улучшений только одна группа может иметь полномочия на обновление модели. Если две разные организации обновляют отдельные модели для одного и того же продукта, контроль над моделью может быть потерян. Последствия этого могут заключаться в том, что изменения, которые, как предполагалось, будут реализованы, не реализуются, или модели «сломаны» и должны быть перестроены — процесс, требующий много времени, который затем необходимо снова проверить на предмет точности.
Изменения в документации
Существует процесс обновления чертежей, процедур и другой документации, используемой в производстве. Этот процесс по-разному называется приказом об инженерных изменениях (ECO), уведомлением об инженерных изменениях (ECN) или аналогичным термином. Дело не в том, что инженеры — единственная группа, которая может обновлять документ, но это группа, которая обновляет большинство документов. Все, что связано с дизайном продукта, спецификациями или процессом, решается инженерно-техническими средствами. Группа качества или руководство могут вносить свои собственные изменения в документацию, но все это делается через систему технических изменений.
Все, что связано с дизайном продукта, спецификациями или процессом, решается инженерно-техническими средствами. Группа качества или руководство могут вносить свои собственные изменения в документацию, но все это делается через систему технических изменений.
Этот процесс также определяет один или несколько шагов утверждения, прежде чем документ может быть выпущен. Это обеспечивает надлежащую проверку документов для выявления ошибок перед выпуском. Неважно, кто пишет или обновляет документ, крайне важно, чтобы еще одна группа специалистов посмотрела на документ перед выпуском, чтобы убедиться, что все изменения были должным образом реализованы или учтены все возможные последствия. В качестве примера рассмотрим продукт с определенным модулем ввода питания. Если продукт имеет нормативную маркировку CE, обычно имеется отчет о нормативном списке, в котором этот компонент документируется как критический компонент для безопасности и, следовательно, не может быть изменен или повторно указан без проверки регулирующими органами. Инженеры могут захотеть изменить компонент по нескольким причинам, но не знать, что отчет о листинге контролирует этот конкретный компонент, и для того, чтобы изменение могло произойти, необходимо выполнить другие действия.
Инженеры могут захотеть изменить компонент по нескольким причинам, но не знать, что отчет о листинге контролирует этот конкретный компонент, и для того, чтобы изменение могло произойти, необходимо выполнить другие действия.
Что такое документооборот в науках о жизни?
В медико-биологической отрасли управление документами означает процессы и стратегии, используемые для управления множеством различных документов, которые перемещаются внутри организации, среди инвесторов и спонсоров, а также регулирующих органов на протяжении всего жизненного цикла продукта.
Документация имеет решающее значение для подтверждения безопасности и эффективности продуктов вашей организации, таких как медицинские устройства и фармацевтические препараты. Документация докажет надежность, воспроизводимость и повторяемость процессов. С помощью документации вы эффективно сообщаете, что должно быть сделано, когда это должно быть сделано и как это должно быть сделано.
Мы рассмотрим следующие темы:
- Определение контроля документов
- Требования к контролю документов
- Процедуры контроля документов
- Важность контроля документов
- Как добиться эффективного контроля документов
- Автоматизация управления документацией
- Часто задаваемые вопросы о контроле документации
Определение контроля документации
Управление документацией можно определить как ряд методов, обеспечивающих создание, проверку, распространение и уничтожение документов организованным и поддающимся проверке образом.
Вы также можете использовать термин «управление документами». Хотя эти термины тесно связаны, они не являются взаимозаменяемыми.
Управление документами указывает на системы и процессы, внедренные в вашей организации для ведения документации и управления ею. Это хранение, расположение, отслеживание, обновление и совместное использование документов.
Давайте рассмотрим несколько примеров из отрасли медико-биологических наук, чтобы лучше понять управление документами.
На производственной линии вашей фармацевтической компании отображается памятка с подробным описанием изменений, которые необходимо внести в существующий производственный процесс. Памятка представляет собой дубликат информации, которая хранится в контролируемых спецификациях в другом месте. Теперь считайте, что в контролируемый документ вносятся дальнейшие изменения. Но служебная записка продолжает содержать устаревшую информацию, потому что она не контролируется. Вы либо начинаете контролировать меморандум, либо уничтожаете его немедленно!
В другом примере отдел обучения вашей компании по производству медицинского оборудования подготовил пересмотренный и обновленный учебный модуль для новых сотрудников. Учитывая важность, придаваемую такому модулю, его необходимо контролировать, а каждое изменение документировать.
Учитывая важность, придаваемую такому модулю, его необходимо контролировать, а каждое изменение документировать.
Рекомендуемая литература
- Руководство по эффективному управлению документооборотом в фармацевтической отрасли
- Документооборот по медицинским изделиям: что это такое и как его упростить
Требования к контролю документации
Поскольку эффективный контроль документации является основой отрасли медико-биологических наук, его регулируют несколько международных руководств и стандартов.
К ним относятся FDA 21 CFR, часть 820.40, ISO 13485:2016, регламент Европейского союза по медицинским устройствам (EU MDR), FDA 21 CFR, часть 11, и GxP.
FDA 21 CFR Part 820.40: Этот стандарт Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США относится к контролю документов для компаний, производящих медицинские устройства, которые производят или продают продукцию на рынке США. Такие компании должны установить и поддерживать процедуры контроля всех соответствующих и сопутствующих документов, которые подчеркнуты в Правилах системы качества (QSR) FDA.
Такие компании должны установить и поддерживать процедуры контроля всех соответствующих и сопутствующих документов, которые подчеркнуты в Правилах системы качества (QSR) FDA.
ISO 13485:2016, раздел 4.2.4 (Контроль документации): Данное руководство Международной организации по стандартизации содержит стандарты для систем управления качеством медицинских изделий (СМК). В отдельном разделе подчеркивается важность контроля документов на протяжении всего жизненного цикла медицинского устройства.
Кроме того, ISO13485:2016 также содержит требование о проверке компьютерной системы (раздел 4.1.6, общие требования), которое актуально для организаций, использующих компьютерное программное обеспечение, такое как eQMS или систему управления документами, для управления своими документами.
Следует иметь в виду, что оба вышеупомянутых стандарта имеют существенные общие черты. Таким образом, когда ваша организация начнет соответствовать одному стандарту, вы также будете готовиться к другому стандарту.
Рекомендуемая литература: Документация по системе менеджмента качества
Основные характеристики для контроля документов в соответствии с ISO 13485:2016 и FDA 21 CFR Part 820
- Организация должна определить наиболее подходящего человека (лиц) для оценки всех процедур относительно соответствующих документов
- Все утверждения контроля документов должны включать подписи утверждающих лиц и дату
- Все применимые точки использования должны иметь актуальную версию документа
- Изменения, если они требуются, должны быть проверены и утверждены одним и тем же лицом( s) которые участвовали в первоначальном рассмотрении и утверждении
- Утвержденные изменения в документах должны быть доведены до сведения всех соответствующих пользователей
EU MDR: Последний регламент по медицинским устройствам, применимый к Европейскому союзу, содержит много моментов, касающихся контроля документации. Акцент делается на жизненном цикле продукта в целом. Это включает в себя послепродажное наблюдение, которое представляет собой набор действий, которые ваша организация будет выполнять для сбора и оценки возможностей, полученных от вашего медицинского устройства, которое уже находится на рынке, и для определения того, нужно ли предпринимать какие-либо дальнейшие действия.
Акцент делается на жизненном цикле продукта в целом. Это включает в себя послепродажное наблюдение, которое представляет собой набор действий, которые ваша организация будет выполнять для сбора и оценки возможностей, полученных от вашего медицинского устройства, которое уже находится на рынке, и для определения того, нужно ли предпринимать какие-либо дальнейшие действия.
FDA 21 CFR Part 11: Системы электронной документации и подписи, вероятно, являются неотъемлемой частью вашей организации, и вам придется соблюдать этот стандарт FDA.
Даже если вы полностью работаете с бумажными документами, но по-прежнему отправляете документы по электронной почте, вам придется соблюдать Свод федеральных правил (CFR) FDA США, часть 11, раздел 21, в котором говорится, что организации, использующие ту или иную форму электронной записи должны:
«защищать созданные компьютером контрольные журналы с отметками времени для независимой записи даты и времени записей оператора, а также действий, которые создают, изменяют или удаляют электронные записи».

Кроме того, FDA 21 CFR Part 11 также содержит особые требования для таких областей, как валидация, создание записей, контрольные журналы, операционные средства контроля и средства контроля безопасности.
GxP: Это сборник правил и руководств по обеспечению качества, гарантирующих, что продукты, производимые медико-биологической отраслью, безопасны, подходят для предполагаемого использования и соблюдают все методы обеспечения качества при их производстве, контроле, хранении и поставке. .
GxP — это аббревиатура от Good (x-переменная в зависимости от применения, например, C: Клинические; L: Лабораторные; M: Производство; S: Хранение; R: Обзор… .) Упражняться.
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США учредило GxP для обеспечения того, чтобы фармацевтические, биотехнологические компании и компании, производящие медицинское оборудование, соответствовали следующим требованиям:
- Подотчетность – способность точно определить лицо (лиц), ответственных за процессы и процедуры
- Прослеживаемость — возможность проследить все процессы от закупки сырья до истории их разработки в качестве медицинского изделия/лекарства
- Целостность данных — возможность полагаться на генерируемые данные
Процедуры контроля документации
Благодаря управлению документами ваша организация будет управлять всеми документами на протяжении всего их жизненного цикла. Это означает, что это надежный, стандартизированный и организованный способ организации документов, данных и информации.
Это означает, что это надежный, стандартизированный и организованный способ организации документов, данных и информации.
Итак, у вас будет набор процедур документооборота и лицо (лица), которое будет отвечать за документооборот. Содержание этих процедур зависит от того, используете ли вы электронную систему для таких задач, как нумерация, управление версиями и архивирование.
Поэтому попросите своего поставщика eQMS предоставить процедуру контроля документов, которая приведена в соответствие с соответствующими правилами, поскольку это сэкономит ваше время.
Ключевые этапы процедуры контроля документов:
- Создание документа
- Рассмотрение и утверждение
- Редакции
- Публикация
- Устаревание/ликвидация
1. Создание документа
эти документы должны быть созданы. Вы также включите сведения о форматировании документа, его названии, истории версий и других технических деталях.
2. Рассмотрение и утверждение документа
Процедура контроля документов в вашей организации также даст подробное объяснение того, как должен быть рассмотрен подготовленный документ и кто является ответственным за его утверждение. Кроме того, в процедуре контроля документов будет выделено, как должны регистрироваться утверждения и модификации. Вы должны знать, что все документы должны быть проверены на точность перед публикацией.
Кроме того, в процедуре контроля документов будет выделено, как должны регистрироваться утверждения и модификации. Вы должны знать, что все документы должны быть проверены на точность перед публикацией.
3. Внесение изменений в документ
Обратите внимание, что даже после того, как документы будут утверждены и доработаны, в будущем в них могут потребоваться изменения. Еще раз, ваша процедура контроля документов выдвинет на первый план процесс пересмотра документов.
Акцент здесь делается на том, кто имеет полномочия инициировать или запрашивать исправления и кто будет выполнять фактические изменения. Процедура также определит методологию идентификации документа в соответствии с его редакцией. Должен быть опубликован только текущий и утвержденный документ, а более старые версии должны быть устаревшими.
4. Публикация документа
После завершения и утверждения документа следующим шагом будет его публикация. Процедура контроля документов вашей организации будет определять параметры, которым необходимо следовать для публикации документа, и лиц, уполномоченных на доступ к нему. Кроме того, ограничения безопасности для опубликованного документа должны быть четко определены.
Кроме того, ограничения безопасности для опубликованного документа должны быть четко определены.
5. Устаревание документа
Это важный шаг в жизненном цикле документа.
Старые версии документа, определяемые по дате или истории версий, удаляются из доступа.
Важность контроля документации
Как только ваша организация усовершенствует процесс контроля документации, вы получите множество преимуществ.
1. Эффективность
У вашей медико-биологической компании будет несколько документов с многочисленными приложениями в разных местах. С помощью контроля документов вы сможете упорядочивать и отслеживать эти документы и следить за тем, чтобы только соответствующие документы размещались в нужных местах.
Например, вы не можете размещать документ, касающийся производственной линии вашего медицинского изделия/лекарства, в месте, доступном для клиентов.
2. Согласованность
Благодаря эффективному документообороту вы избавлены от многих хлопот и головной боли!
Все ваши сотрудники будут работать по одним и тем же актуальным документам и стандартам.
Предположим, что ваша организация инициировала политику оплачиваемого отпуска по уходу за ребенком для всех сотрудников, имеющих на это право. Однако соответствующие документы опубликованы не во всех ведомствах. Недовольные сотрудники поднимут шум и крик.
3. Качество
Должна быть опубликована самая последняя версия всех документов, а более старые версии должны быть устаревшими.
Например, ваши постмаркетинговые исследования показали, что после небольшой настройки ваш пульсоксиметр будет работать лучше. Но если у производственной группы нет соответствующего документа, вы все равно будете продавать менее эффективный пульсоксиметр.
Аналогичным образом, на основании информации от клиницистов, ваша фармацевтическая компания изменила дозировку жизненно необходимого лекарства. Вам все еще необходимо пересмотреть соответствующие документы и поставить в известность об этом соответствующий персонал.
4. Соответствие
Ваша организация должна будет соблюдать все соответствующие национальные и международные стандарты/рекомендации, упомянутые в предыдущем разделе. Для этого необходимо иметь возможность предъявлять соответствующие документы по требованию. Не может быть оправданий по поводу утерянных документов, неактуальных документов и так далее.
Для этого необходимо иметь возможность предъявлять соответствующие документы по требованию. Не может быть оправданий по поводу утерянных документов, неактуальных документов и так далее.
5. Безопасность
Возьмем гипотетический сценарий недовольного сотрудника ключевого отдела. Человек может получить доступ к конфиденциальным документам и отправить их по электронной почте или нести информацию на электронном устройстве.
Также нам следует опасаться внешних хакеров. Вы можете предотвратить это, установив протоколы безопасности в вашей системе управления документами, чтобы исключить несанкционированный доступ к конфиденциальной информации.
6. Доступность
При хорошем контроле документации ваша организация гарантирует, что каждый отдельный документ и все версии будут легко доступны.
Например, возникла проблема с новым этапом жизненного цикла вашего медицинского устройства или фармацевтического продукта. Если доступны предыдущие версии документов, вы можете отслеживать каждый шаг производственного цикла, выявлять проблемы и устранять их.
7. Повышенная оперативность
Когда организации осознают необходимость хорошего управления документами и предпринимают шаги для его достижения, они становятся активными.
Они могут быть на шаг впереди правил, а не возиться с режимом контроля повреждений.
Как обеспечить эффективный контроль документации
Теперь, когда вы знаете, что такое контроль документации, давайте рассмотрим некоторые из наиболее важных аспектов эффективного контроля документации.
1. Нанять контролера документов
Контролер документов/специалист по контролю документов является лицом, ответственным за все процедуры контроля документов. Если ваша организация среднего или крупного размера, у вас может быть более одного контролера документов. Поскольку эта роль требует технических знаний более чем в одной области, рекомендуется иметь несколько контролеров документов.
2. Выберите правильное программное обеспечение для управления документами
При правильном программном обеспечении для управления документами ваша организация получит преимущество перед конкурентами.
Такое программное обеспечение может безопасно хранить все важные документы в облаке и должно иметь инструменты для организации данных и возможности удобного поиска, навигации.
Программное обеспечение должно поддерживать различные типы документов, включая PDF-файлы, документы Word, файлы изображений, электронные таблицы и другие. Еще один важный аспект, который вам необходимо учитывать, заключается в том, что программное обеспечение должно полностью соответствовать требованиям FDA 21 CFR, часть 11.
Еще одно важное требование для большинства медико-биологических организаций заключается в том, что программное обеспечение для управления документами должно пройти валидацию компьютерной системы — в соответствии с FDA 21 CFR. Часть 11 и, наконец, ISO 13485:2016.
3. Наличие процедуры контроля документации
Как уже упоминалось в подразделе «Процедуры контроля документации», ваша организация должна иметь надлежащее руководство по процедурам контроля документации для достижения эффективного контроля документации.
Поэтому обязательно узнайте у поставщика программного обеспечения для контроля документов, могут ли они предоставить этот документ.
4. Необходимо постоянное совершенствование
По мере того, как ваша организация будет укрепляться, вам потребуется улучшать и обновлять управление документооборотом.
Процесс восстановления может показаться пугающим, но, разбив каждый процесс на управляемые задачи, вы сможете достичь его легче, чем вы думали.
Что такое программное обеспечение для управления документами
Программное обеспечение для управления документами позволяет вашей организации автоматизировать записи всех политик управления, регулировать доступ к конфиденциальной информации и получать полную информацию о деятельности всех документов для аудита и представления.
Когда вы замените бумажные процедуры контроля документов программным обеспечением электронного контроля документов (СЭД), вы получите систему, в которой гарантировано качество и соответствие требованиям. С помощью такой EDMS вы можете управлять информацией и файлами в соответствии с правилами и стандартами, такими как FDA и ISO.
С помощью такой EDMS вы можете управлять информацией и файлами в соответствии с правилами и стандартами, такими как FDA и ISO.
В случае отрасли наук о жизни EDMS обычно называют электронной системой управления качеством (eQMS). EQMS помогает вам управлять всеми вашими процессами QMS.
Давайте рассмотрим пару примеров из отрасли наук о жизни, чтобы лучше понять это.
Из-за нынешней пандемии фармацевтическая компания сталкивается с нехваткой персонала на месте. Однако это не проблема, поскольку их eQMS обеспечивает удаленный доступ через веб-приложение в режиме 24/7.
Ваша компания, производящая медицинские устройства, сталкивается с проблемами, связанными с определенными спецификациями, которые должны быть включены в новое медицинское устройство. С помощью eQMS вы можете регистрировать проблемы качества, с которыми вы сталкиваетесь в разных отделах, и легко их решать.
С электронной системой управления качеством, такой как SimplerQMS, вы не только будете уверенно управлять всеми своими процессами системы управления качеством, но также получите расширенные возможности для принятия решений, наглядность и готовность к требованиям FDA 21 CFR Part 820, ISO 13485:2016, EU MDR , FDA 21 CFR часть 11 и GxP.
Решение SimplerQMS eQMS обеспечивает беспрепятственное сотрудничество между вашей организацией и другими организациями (поставщиками, клиентами и регулирующими органами), лучшую стандартизацию, упрощенную обработку проверки и утверждения, а также эффективное взаимодействие с Microsoft Office Suite.
Рекомендуемая литература: Как выбрать программное обеспечение для управления качеством?
Автоматизация управления документооборотом
С помощью eQMS, такой как SimplerQMS, можно благотворно повлиять на несколько этапов управления документооборотом.
1. Создание и маршрутизация документов
Автоматизация помогает различать документы, такие как документы политики и документы проекта.
Затем им назначается их рабочий процесс, маршрутизация, проверка и утверждение. Поэтому документ рассматривается и утверждается нужными людьми.
2. Контроль версий документов
Автоматизация поможет в процессе запроса на изменение, что гарантирует, что все изменения версии документа соответствуют процессам, определенным вашей организацией.
Программное обеспечение для управления документами будет гарантировать, что все изменения, связанные с данным документом, будут внесены в единый рабочий процесс. Кроме того, старые версии документа автоматически архивируются.
3. Уведомления
Все внутренние и внешние пользователи автоматически получают оповещения, когда необходимая информация поступает/обрабатывается и наступает время действовать.
4. Резервное копирование документов и аварийное восстановление
Резервное копирование данных выполняется автоматически каждые 15 минут, хранится в нескольких местах по всему миру и круглосуточно контролируется профессионалами.
Вы избегаете физического хранения этих документов в своей организации, и у вас есть готовый страховой полис на случай потери или повреждения этих записей.
Часто задаваемые вопросы о контроле документов
Как запустить контроль документов?
В качестве первого шага определите все документы в вашей организации.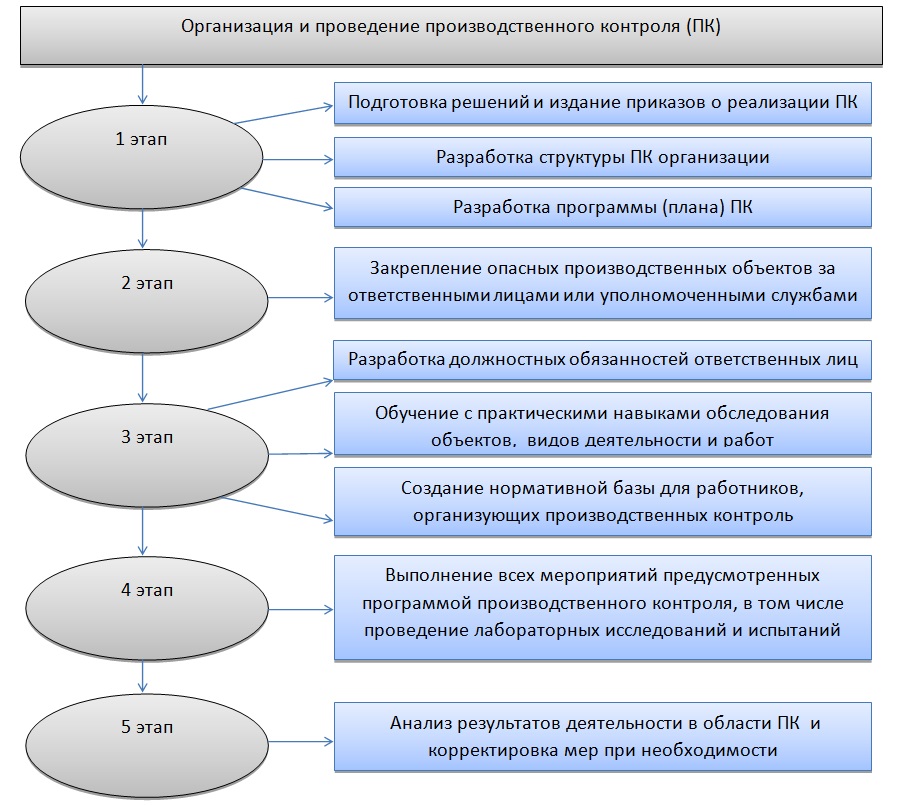 Далее вы установите право собственности и стандарты качества для этих документов. Затем вы систематически именуете и классифицируете документы для надлежащего хранения и поиска. Вам также потребуется создать протоколы ревизий и регулировать доступ и безопасность этих документов.
Далее вы установите право собственности и стандарты качества для этих документов. Затем вы систематически именуете и классифицируете документы для надлежащего хранения и поиска. Вам также потребуется создать протоколы ревизий и регулировать доступ и безопасность этих документов.
Чем занимается сотрудник по контролю документов?
Контролер документов/специалист по контролю документов несет ответственность за все процедуры контроля документов. Такой человек нужен только при наличии бумажной системы документооборота. Если вы вложили средства в EDMS/eQMS, контроллер документов становится излишним.
Зачем нужен документооборот?
Информация является источником жизненной силы всех организаций, будь то науки о жизни или другие. Для эффективного функционирования и регулирования необходим надлежащий поток информации. Такая информация является актуальной, точной, доступной и соответствует характеру вашего бизнеса с надлежащими протоколами и процедурами контроля документов.
Что такое контроль версий документов?
Управление версиями документа — это процесс, с помощью которого вы будете отслеживать различные версии данного документа и управлять ими. Вы будете знать, какая последняя итерация данного документа.
Заключительные мысли
Работая в сфере медико-биологических наук, вы будете иметь дело с информацией изо дня в день. Эта информация должна иметь надлежащий поток, быть точной, доступной, защищенной и актуальной. Все это возможно при наличии надлежащих протоколов и процедур контроля документов.
С помощью eQMS, такой как SimplerQMS, ваша организация не только будет уверенно управлять всеми процессами QMS, но и у вас будут расширенные возможности для принятия решений, наглядность и готовность к национальным и международным нормативным стандартам, таким как FDA 21 CFR Part 820.40, FDA 21 CFR, часть 11, ISO 13485:2016, EU MDR и GxP.
Что такое система контроля документов? — QMS
Часто повторяемая поговорка «если это не задокументировано, то этого не было» навсегда запечатлелась в умах менеджеров по качеству во всем мире. Они поставили контроль документации в основе управления качеством — ни один процесс не начинается, не завершается, не пересматривается, не повторяется или не заменяется без тщательного документирования.
Они поставили контроль документации в основе управления качеством — ни один процесс не начинается, не завершается, не пересматривается, не повторяется или не заменяется без тщательного документирования.
Также можно сказать, что если это задокументировано, но документы не могут быть найдены, значит, этого не было. В медико-биологической отрасли такой надзор может привести к несоблюдению требований и дорогостоящим задержкам с выпуском продукта на рынок.
Документы и записи служат доказательством того, что ваша компания придерживается надлежащих методов производства и управления документами, необходимых для обеспечения качества и соответствия нормативным требованиям. Система управления документами — это процесс организации, маршрутизации, отслеживания, авторизации и распространения всей документации, связанной с проектированием, разработкой и производством продуктов. Контроль документации также продолжается в процессах послепродажного надзора для отслеживания документации, касающейся жалоб и корректирующих и предупреждающих действий (CAPA).
Типы документации, необходимой для разработки подкарантинной продукции
Медико-биологические компании выпускают значительный объем документации, большая часть которой должна быть доступна аудиторам во время проверок. Ниже приведены некоторые документы, сопровождающие разработку продукта:
- Стандартные операционные процедуры (СОП).
- Руководства по качеству.
- Оформление входных и выходных документов.
- Планы испытаний.
- Контрольные списки проверки.
- Журналы несоответствий.
- Журналы отклонений.
- отчетов CAPA.
Тщательное обращение с большими объемами документации является сложной задачей, но крайне важно, чтобы все документы были полными, точными, актуальными и включали все метаданные. Согласно 21 CFR 820, каждый контролируемый документ должен четко указывать:
- Кто создал документ.
- Кто проверил документ и данные.
- Кто утвердил документ и когда.
- Статус документа (действителен, черновик, в доработке, ожидает утверждения).
- Номер редакции документа.
- Авторизация (контролируемая/зарегистрированная копия или авторизованная распечатка с подписями).

Контроль документов выходит за рамки флажков
Процесс документирования, который следует вышеупомянутым рекомендациям, соответствует базовому (контрольному списку) стандарту системы контроля документов. После этого компании обычно создают собственную структуру и процессы для управления документацией, используя бумажные, электронные или комбинацию обоих типов инструментов, чтобы соответствовать требованиям соответствия.
Использование бумажной или гибридной системы, которая придерживается нормативных требований ради соответствия, может неблагоприятно повлиять на сроки производства и время выхода на рынок. Механика внесения изменений, направление документов на утверждение и поиск или переделывание утерянных документов — самые большие препятствия в жизненном цикле продукта. Рассмотрим следующий «гипотетический» сценарий бумажной системы документооборота.
Рассмотрим следующий «гипотетический» сценарий бумажной системы документооборота.
Управление документооборотом (не так) Передовой опыт
Вы идете по производственному участку с менеджером по качеству компании, чтобы увидеть их систему контроля документации в действии. Вы случайно выбрали процедурный документ, который включает процедуры настройки, параметры калибровки, критерии приемки и рабочие инструкции. Документ, который вы носите, тщательно отформатирован, распечатан и аккуратно помещен в папку.
Вы подходите к рабочей станции и видите тот же документ. Сразу же у вас есть довольно хорошее представление о том, как работает их система контроля документов, которая, кажется, включает в себя экономию денег на бумаге и печати. Процессуальный документ представляет собой заверенную фотокопию, скрепленную большой скрепкой, которая с трудом удерживает все страницы на месте. На первой странице указан номер редакции документа, а также дата, нацарапанная под предыдущей редакцией, и дата, обе из которых зачеркнуты. Существует рукописный список инструкций, где обновленные разделы должны заменить устаревшие разделы. В нижней части первой страницы вы замечаете ряд подписей, которые, как вы интуитивно понимаете, являются автографами лиц, утверждающих документ.
Существует рукописный список инструкций, где обновленные разделы должны заменить устаревшие разделы. В нижней части первой страницы вы замечаете ряд подписей, которые, как вы интуитивно понимаете, являются автографами лиц, утверждающих документ.
На другой рабочей станции вы видите более старую версию того же документа, вырезанного из папки, только без обновленных инструкций. Вы предполагаете, что обновленные инструкции еще не поступили, а затем обнаруживаете, что это предположение неверно. Пролистывая документ, вы видите зачеркнутые разделы содержания и примечания, сопровождаемые подписями, нацарапанными на полях.
Похоже, другой человек ускорил процесс внесения изменений в этот документ. Вместо того, чтобы распечатывать и маршрутизировать обновленные разделы, этот человек мог бы упростить процесс маршрутизации, обходя каждого заинтересованного лица и ожидая, пока каждый человек вручную введет и подпишет исправления. Затем этот же документ просто возвращался на рабочую станцию. Наличие нескольких активных версий одного и того же документа затрудняет определение правильного черновика, что аннулирует аспект управления версиями в управлении документами.
Наличие нескольких активных версий одного и того же документа затрудняет определение правильного черновика, что аннулирует аспект управления версиями в управлении документами.
Следующей остановкой вашего тура будет складское помещение, в котором хранятся стопки коробок, служащих хранилищем документации. На каждой коробке есть этикетка, показывающая тип и статус хранящихся в ней документов. Сотрудники по контролю документов отслеживают и находят всю документацию, используя распечатанные электронные таблицы, прикрепленные к буферу обмена. Чтобы получить запрошенную версию документа, сотрудники просеивают ящики, чтобы найти то, что им нужно.
Эффективное управление документами — сложная задача для бумажной системы
Хотя ранее описанный «процесс» документации соответствует нормативным требованиям, он не совсем пример эффективной, масштабируемой системы управления документами. Являясь центральной частью общей картины управления качеством, документация оказывает большое влияние на всю операцию. Когда возникает конфликт точности данных, утверждающий недоступен или документ теряется, задержки производства практически гарантированы.
Когда возникает конфликт точности данных, утверждающий недоступен или документ теряется, задержки производства практически гарантированы.
С таким количеством движущихся частей бумажная система управления документами подвержена большому количеству ошибок и несоответствий. Многие организации переходят на оцифрованные системы управления документами, чтобы устранить нарушения, связанные с документацией, и сбои в производстве.
Помимо предотвращения задержек в маршрутизации и потери документов, электронная система позволяет нескольким заинтересованным сторонам одновременно просматривать документ, устранять конфликты и ставить подписи для утверждения из любого места. Система управления документацией , которая интегрируется со всеми другими компонентами системы качества, автоматизирует маршрутизацию и распределение, отправляет напоминания заинтересованным сторонам и предупреждает необходимый персонал о необходимости обучения новым процедурам. Ваша система документооборота должна ускорять, а не мешать продвижению продукта на рынок.
Дэвид Дженсен — специалист по контент-маркетингу в MasterControl, где он отвечает за исследование и написание контента для веб-страниц, официальных документов, брошюр, электронных писем, сообщений в блогах, презентационных материалов и социальных сетей. Он имеет более чем 25-летний опыт создания учебного, маркетингового контента и материалов по связям с общественностью для различных отраслей и аудиторий, связанных с технологиями. Дженсен много пишет о кибербезопасности, целостности данных, облачных вычислениях и производстве медицинского оборудования. Он публиковал статьи в различных отраслевых изданиях, таких как Medical Product Outsourcing (MPO) и Bio Utah. Дженсен имеет степень бакалавра коммуникаций Государственного университета Вебера и степень магистра профессиональных коммуникаций Вестминстерского колледжа.
Управление документами и изменениями в процессе проектирования
Промышленный журнал медицинского оборудования и диагностики
Указатель статей MDDI
Первоначально опубликовано в апреле 2000 г.
При правильном применении управление документированием и изменениями снижает количество дефектов продукции, сводит к минимуму проблемы на местах и повышает удовлетворенность конечных пользователей.
01 апреля 2000 г.
Медицинское оборудование и диагностическая промышленность Журнал
Указатель статей MDDI
Первоначально опубликовано в апреле 2000 г.
При правильном применении управление документированием и изменениями снижает количество дефектов продукции, сводит к минимуму проблемы на местах и повышает удовлетворенность конечных пользователей.
Контроль документации начинается на ранней стадии процесса проектирования и распространяется не только на первоначальный выпуск проекта, но и на весь жизненный цикл продукта. Проектно-выходные документы составляют основу основной записи устройства (DMR), которая в конечном итоге передается в производство. Документирование и управление изменениями помогают преодолеть разрыв между контролем производства и проектирования.
Документирование и управление изменениями помогают преодолеть разрыв между контролем производства и проектирования.
Дисциплина и коммуникация являются ключом к эффективному документированию и системам управления изменениями. Знание того, как принимать важные решения, касающиеся изменений в конструкции и производстве, и понимание того, какие средства контроля следует применять для каждого из них, являются жизненно важными навыками для производителей. В данной статье рассматриваются нормативные требования и обсуждаются способы внедрения систем документооборота. Он охватывает основы создания и обслуживания систем для контроля за просмотром, утверждением и распространением документов, а также объясняет, как интегрировать текущий контроль изменений проекта с системами для контроля за документами и изменениями.
НОРМАТИВНЫЕ ТРЕБОВАНИЯ
Контроль документации является важным элементом эффективной системы качества. Документы демонстрируют, что системы и средства контроля, используемые для обеспечения того, чтобы готовые устройства соответствовали спецификациям, были эффективно внедрены. Они сообщают о желаемых результатах, разъясняют методы и обеспечивают согласованные результаты. При правильном применении контроль документов помогает снизить вероятность дефектов и свести к минимуму проблемы на местах. В конечном итоге эти улучшения повышают удовлетворенность клиентов.
Они сообщают о желаемых результатах, разъясняют методы и обеспечивают согласованные результаты. При правильном применении контроль документов помогает снизить вероятность дефектов и свести к минимуму проблемы на местах. В конечном итоге эти улучшения повышают удовлетворенность клиентов.
Требования к контролю документов определены в разделе 820.40, подраздел D, регламента системы качества (QSR) для медицинских устройств. 1 Общие требования заключаются в том, чтобы «установить и поддерживать процедуры для контроля всех документов, требуемых этой частью». В этом случае «все» можно разбить на две основные категории: процедуры систем качества и документы DMR (см. Таблицу I). Во время проектирования и разработки интересующие документы составляют значительную часть файла истории проектирования, который в конечном итоге будет передан в DMR. Он включает в себя как печатные, так и электронные форматы.
| Процедуры систем качества | Основная запись устройства/файл истории разработки |
|---|---|
| Руководства | Спецификации и чертежи |
| СОП, рабочие инструкции | Технологическое оборудование и средства управления |
| Формы | Процедуры проверки и испытаний |
| Списки утвержденных поставщиков | Критерии приемки |
| Заказы на поставку | Упаковка и маркировка |
| Процедуры установки, технического обслуживания и обслуживания |
 Образцы документов для контроля.
Образцы документов для контроля. Все документы должны быть проверены на адекватность и утверждены перед распространением. Утверждения должны быть задокументированы, а средства контроля должны гарантировать, что нужные документы используются в нужном месте. Во многих случаях правый документ является самой последней редакцией или выпуском. Это означает, что старые или устаревшие версии должны быть удалены из мест использования. Если для создания запасных частей необходимо поддерживать старые (устаревшие) версии, то следует установить средства контроля, чтобы предотвратить случайное использование неправильного документа и гарантировать, что используется правильная версия.
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) признает электронные подписи на документах, если установлены соответствующие меры контроля, гарантирующие, что каждая электронная метка или идентификация может быть выдана только лицом, уполномоченным использовать метку и утверждать документ. 2 Электронные документы должны контролироваться так же, как и бумажные документы, включая защиту от несанкционированных изменений.
2 Электронные документы должны контролироваться так же, как и бумажные документы, включая защиту от несанкционированных изменений.
Когда в документы вносятся изменения, они должны быть рассмотрены и утверждены с использованием тех же методов, которые использовались для первоначального рассмотрения документов, если другой метод не может быть оправдан. Это помогает гарантировать, что документ будет иметь тот же уровень проверки, что и изначально. Кроме того, должны быть предусмотрены условия для сообщения об изменениях соответствующему персоналу и ведения записей об изменениях.
Несколько других разделов QSR также относятся к контролю документов. Оба раздела для данных о закупках (820.50(b)) и производственных и технологических изменений (820.70(b)) управляют справочным документом (820.40). Кроме того, в нормативных актах переплетаются элементы контроля за дизайном и документацией. Подчасть C, элементы управления проектированием (раздел 820.30(h), передача проекта), требует от производителей «обеспечить, чтобы проектная основа устройства была правильно переведена в производственные методы и процедуры». Как правило, последним шагом в этом процессе является рассмотрение и утверждение DMR.
Как правило, последним шагом в этом процессе является рассмотрение и утверждение DMR.
Наконец, раздел об изменениях в проекте, 820.30(i), требует, чтобы изменения в проекте были идентифицированы, задокументированы, подтверждены (или проверены, если необходимо), проверены и одобрены до внедрения. Для компаний типично иметь две системы: одну систему с более простыми элементами управления для управления конфигурацией проекта во время проектирования и разработки, а другую для управления процедурами системы качества и документами DMR. 3 Однако можно использовать одну и ту же систему в течение всего процесса проектирования.
ПОЛУЧЕНИЕ КОНТРОЛЯ ДОКУМЕНТОВ
Первый вопрос, который часто задают производители, заключается в том, когда в процессе проектирования они должны инициировать контроль документов. Даже в начинающих компаниях, где производители разрабатывают свой первый продукт, необходимо руководство по системам качества. Как только принято решение о разработке продукта для рынка, должны быть введены в действие подробные процедуры контроля дизайна. Кроме того, должны быть четко определены вспомогательные системы, такие как ответственность руководства, обучение, контроль закупок, контроль калибровки и, конечно же, контроль документации. Как только процесс управления проектированием начинает генерировать результаты проектирования, необходимо контролировать все документы (см. рис. 1). Кроме того, результаты любого этапа проектирования должны быть рассмотрены и одобрены. Сюда входят проектные входные и выходные документы. Если вносятся изменения, применяются требования по управлению изменениями проекта. Запись исправлений недостатков проекта является важной частью файла истории проекта и помогает предотвратить подобные ошибки при будущих изменениях. 4 Любые обзоры проекта, анализы рисков, проверки или валидации должны обновляться или повторяться, чтобы гарантировать, что выходные данные проекта соответствуют входным данным проекта, а спецификации устройства последовательно соответствуют потребностям пользователя и предполагаемому использованию.
Кроме того, должны быть четко определены вспомогательные системы, такие как ответственность руководства, обучение, контроль закупок, контроль калибровки и, конечно же, контроль документации. Как только процесс управления проектированием начинает генерировать результаты проектирования, необходимо контролировать все документы (см. рис. 1). Кроме того, результаты любого этапа проектирования должны быть рассмотрены и одобрены. Сюда входят проектные входные и выходные документы. Если вносятся изменения, применяются требования по управлению изменениями проекта. Запись исправлений недостатков проекта является важной частью файла истории проекта и помогает предотвратить подобные ошибки при будущих изменениях. 4 Любые обзоры проекта, анализы рисков, проверки или валидации должны обновляться или повторяться, чтобы гарантировать, что выходные данные проекта соответствуют входным данным проекта, а спецификации устройства последовательно соответствуют потребностям пользователя и предполагаемому использованию.
|
На рис. 1 показано, что по мере того, как проект передается и преобразуется в DMR, документы должны быть проверены и утверждены, чтобы убедиться, что проект правильно переведен в процедуры и спецификации для производства. Как правило, это не происходит сразу, а происходит на протяжении всего процесса разработки по мере завершения элементов дизайна.
Как правило, это не происходит сразу, а происходит на протяжении всего процесса разработки по мере завершения элементов дизайна.
Рисунок 2 дает несколько советов о том, что следует учитывать при проверке адекватности документа DMR. Список не является исчерпывающим, и каждый пункт может не относиться к каждому документу, но контрольный список обеспечивает общую отправную точку для проверки документов. Проверка документов может выполняться как часть окончательного утверждения или до окончательного утверждения. Если проверка выполняется как часть окончательного утверждения путем направления документа на утверждение, производители должны быть осторожны, чтобы получить повторное утверждение, если в процессе утверждения будут внесены изменения. По этой причине во многих организациях проводятся обзорные и заключительные собрания для сбора всех входных данных во время окончательного обзора; там по мере необходимости вносятся изменения и документируется окончательное утверждение.
Рисунок 2. Контрольный список проверки документов DMR.
Контрольный список проверки документов DMR.
Производители должны решить, какие органы утверждения будут лучше всего работать в их организации. Важно следить за тем, как работает процесс проверки и утверждения, и при необходимости изменять его. Например, если требуются частые заказы на изменение для исправления элементов, пропущенных в процессе проверки и утверждения, то следует определить основные причины и внести необходимые улучшения.
СМЕНА КОНТРОЛЯ
Определить изменение просто. Любое изменение существующего утвержденного документа считается изменением документа. К движущим силам изменений могут относиться:
- Проблемы с качеством в результате корректирующих и предупреждающих действий.
- Маркетинг или потребности клиентов.
- Нормативные или стандартные изменения.
- Повышение производительности или затрат.
- Изменения поставщика или сырья.
- Усовершенствования документа или использование другого формата.

Существуют две основные области изменений: дизайн и процесс. Изменение конструкции – это изменение конструктивной основы устройства. Стимулом для изменения дизайна часто являются полевые или другие проблемы с качеством. Изменения процесса изменяют методы управления процессом, но не предназначены для изменения конструкции; однако изменение конструкции может вызвать изменения в процессе. Имея дело с изменениями конструкции, производителям необходимо решить, какие типы средств контроля конструкции следует применять.
На рис. 3 показан процесс принятия решения о том, какие типы управления изменениями необходимы. Если изменение относится только к процессу, для реализации можно использовать средства управления производственными изменениями. Если, с другой стороны, изменение связано с конструкцией, первое определение, которое необходимо сделать, должно заключаться в том, будет ли предполагаемое использование устройства таким же. Если это не так, производитель должен начать процесс проектирования с самого начала. Все шаги не обязательно повторять полностью, но каждый из них должен быть по крайней мере рассмотрен и определен как ненужный, прежде чем двигаться дальше. Блок-схема (рис. 3) может отличаться в зависимости от конкретного процесса проектирования, но она дает рекомендации для принятия решений. Кроме того, производители также должны определить, требуется ли подача предпродажного уведомления 510(k) или дополнения к предварительному утверждению. Решения, принятые с использованием блок-схемы на рис. 3, могут помочь в подтверждении этих решений о представлении.
Все шаги не обязательно повторять полностью, но каждый из них должен быть по крайней мере рассмотрен и определен как ненужный, прежде чем двигаться дальше. Блок-схема (рис. 3) может отличаться в зависимости от конкретного процесса проектирования, но она дает рекомендации для принятия решений. Кроме того, производители также должны определить, требуется ли подача предпродажного уведомления 510(k) или дополнения к предварительному утверждению. Решения, принятые с использованием блок-схемы на рис. 3, могут помочь в подтверждении этих решений о представлении.
Рисунок 3. Решения по управлению изменениями.
Кроме того, должен быть установлен какой-то механизм, помогающий облегчить принятие решений по управлению изменениями. Контрольный список, например, может помочь отфильтровать некоторые из более рутинных изменений, которые происходят на постоянной основе, например, исправления или улучшения документа. Анализ рисков также можно использовать для определения соответствующего уровня проектных средств контроля. Как только производители решили, с чего начать процесс проектирования, они могут применять соответствующие процедуры контроля проектирования.
Как только производители решили, с чего начать процесс проектирования, они могут применять соответствующие процедуры контроля проектирования.
Простые изменения конструкции с низким уровнем риска могут быть обработаны с соответствующей проверкой, валидацией и анализом конструкции. Сложные и сопряженные с повышенным риском изменения следует планировать более тщательно, а также выполнять более обширную проверку, валидацию и анализ проекта. Важно не только учитывать требования к проверке и валидации предлагаемых изменений, но и определять возможные неблагоприятные последствия для других функций устройства. В любом случае члены кросс-функциональной команды должны проанализировать эти решения, чтобы убедиться, что реализованы соответствующие элементы управления.
Производители также должны решить, требует ли изменение пересмотра существующего документа или требует создания нового документа и номера детали (или номера документа). Это решение требует тщательного рассмотрения, поскольку изменения могут повлиять на другие документы и могут потребовать дорогостоящих изменений в системе планирования производства. Обычные критерии принятия решений включают оценку того, меняет ли изменение форму, соответствие или функцию. Если какой-либо из этих критериев изменится, то может потребоваться новый номер детали или документа.
Обычные критерии принятия решений включают оценку того, меняет ли изменение форму, соответствие или функцию. Если какой-либо из этих критериев изменится, то может потребоваться новый номер детали или документа.
По сути, производители решают, взаимозаменяемы ли старое и новое. Иногда это практический вопрос: если старая версия не работала и больше не нужна, то изменение номера детали поможет предотвратить ошибки. Если изменение повлекло за собой изменение формы, посадки или функции, изготовителю, возможно, также придется рассмотреть варианты модернизации существующих устройств.
КОНТРОЛЬ ПРОИЗВОДСТВЕННЫХ ИЗМЕНЕНИЙ
Часто изменения конструкции обрабатываются с использованием процедур контроля производственных изменений. Это происходит либо потому, что в процесс вносятся изменения, либо потому, что изготовитель хочет облегчить передачу изменения конструкции в производство. Процедуры управления производственными изменениями обеспечивают системы не только для контроля и утверждения, но и для оценки изменений и их интеграции в нормальный производственный процесс. Факторы, которые необходимо учитывать при разработке системы управления производственными изменениями, включают:
Факторы, которые необходимо учитывать при разработке системы управления производственными изменениями, включают:
- Необходимость проверки процесса.
- Заводские испытания или пилотные прогоны.
- Размещение тестовых материалов.
- Эффективность изменений и отслеживаемость.
- Утилизация устаревшего инвентаря.
- Общение и обучение.
Эти вопросы необходимо решить на ранней стадии процесса изменений. Ожидание более поздних этапов процесса может привести к задержке утверждения, поскольку проблемы должны быть решены к удовлетворению утверждающих лиц. Иногда создается система запросов на изменение, чтобы заблаговременно планировать и санкционировать изменение. В этом типе системы запрошенное изменение и последующий план контроля реализации предлагаются, рассматриваются и утверждаются. План может включать соответствующие элементы управления проектированием, валидацию процесса и другие аспекты реализации. После утверждения план реализуется. Если все критерии для изменения соблюдены, изменение реализуется с использованием запроса на изменение.
Если все критерии для изменения соблюдены, изменение реализуется с использованием запроса на изменение.
Ключом к ответу на проверочные вопросы является понимание того, как был изменен процесс. Если процесс новый, главный вопрос, можно ли его полностью проверить последующими проверками и испытаниями? Если нет, он должен быть подтвержден. Если процесс изменен и ранее не был подтвержден, применяется тот же вопрос. Ранее проверенные процессы требуют повторной проверки или, по крайней мере, обоснования того, почему существующая проверка поддерживает изменения. Однако, если происходят отклонения, производителям необходимо рассмотреть вопрос о повторной аттестации. Например, если в процесс вносятся изменения из-за того, что количество дефектов слишком велико, скажем, производительность составила всего 95 %, в то время как предыдущая валидация хвасталась выходом процесса в 99,9 % — тогда старая валидация, вероятно, была неадекватной и необходима повторная валидация. Использование инструментов проверки процесса также может помочь установить эффективность изменений процесса, в отличие от использования производственной среды в качестве испытательного полигона.
Валидация процесса или другие квалификационные исследования существующего процесса могут быть выполнены с использованием заводских испытаний или пилотных процедур. Поскольку это обычно делается в процессе производства, а изменения не были рассмотрены и утверждены, для контроля документации прототипа и любого произведенного продукта требуется временное уведомление об изменении. Продукт, созданный во время тестовых прогонов, должен быть помещен на карантин до завершения всех испытаний и получения окончательных оценок и утверждений. Общее правило для окончательного размещения продукта, сделанного во время тестового запуска, заключается в том, чтобы задать вопрос: изменилась ли окончательная конфигурация документов после запуска теста? Если ответ отрицательный, то после того, как изменение будет официально рассмотрено и одобрено, продукт можно будет использовать. Если конфигурация изменилась, производитель должен решить, насколько существенными были изменения, а затем иметь возможность обосновать выпуск любого произведенного продукта. Если обоснование невозможно, требуется повторная валидация.
Если обоснование невозможно, требуется повторная валидация.
Решая, в какой момент вводить изменения в производство, производители должны принимать во внимание потребность в изменении, запасы старого материала и доступность нового материала. Если старый дизайн неприемлем из-за риска, связанного с продуктом, изменение должно быть инициировано немедленно, а старый инвентарь списан или переработан. В противном случае производитель должен сбалансировать экономику переработки старых запасов с преимуществами сокращения замены и доступности новых деталей.
Вопросы коммуникации и обучения жизненно важны для успеха изменений. Во многих случаях необходимо тщательно отслеживать изменения, чтобы свести к минимуму влияние проблем на производство. Операторам часто необходимо пройти переподготовку, чтобы внедрить изменения в процесс.
ЗАКЛЮЧЕНИЕ
Начало контроля документов на ранних стадиях проектирования и разработки помогает предотвратить проблемы в будущем. Эти элементы управления применяются как к процедурам систем качества, так и к документам DMR. Контроль проверки и утверждения должен помочь предотвратить выпуск неточных или несоответствующих документов. Средства управления распространением должны быть простыми, видимыми и обеспечивать использование только правильных документов. Средства управления изменениями должны быть сосредоточены на планировании, проверке (или проверке) и внедрении изменений, чтобы свести к минимуму потенциальные проблемы с качеством. Ключами к эффективному документированию и управлению изменениями являются:
Эти элементы управления применяются как к процедурам систем качества, так и к документам DMR. Контроль проверки и утверждения должен помочь предотвратить выпуск неточных или несоответствующих документов. Средства управления распространением должны быть простыми, видимыми и обеспечивать использование только правильных документов. Средства управления изменениями должны быть сосредоточены на планировании, проверке (или проверке) и внедрении изменений, чтобы свести к минимуму потенциальные проблемы с качеством. Ключами к эффективному документированию и управлению изменениями являются:
- Четко определенный процесс.
- Внимание к деталям.
- Связь.
- Дисциплина.
- Валидация или проверка изменений перед внедрением.
ССЫЛКИ
Свод федеральных правил , 21 CFR 808, 812 и 820.
Свод федеральных правил , 21 CFR, часть 11.
Руководство по контролю конструкции для производителей медицинских изделий , (Роквилл, Мэриленд: Центр устройств и радиологического здоровья, FDA, 11 марта 1997 г.